- The Road to 2026: What the New EDS Classification Update Means for You - 30 March 2026
- Parsonage-Turner Syndrome and COVID-19: What the Research Shows - 28 March 2026
- Managing Pain in the Acute Phase of Parsonage-Turner Syndrome - 28 March 2026
If you’ve been diagnosed with hypermobile Ehlers-Danlos syndrome, or if you’ve spent years trying to get that diagnosis, you’ll know how much the 2017 classification meant to this community. For the first time, there was an internationally agreed set of criteria. There was a name, hEDS, that carried real clinical weight. And there was a framework that, in theory, should have made it easier for doctors to understand what they were dealing with.
The problem is, theory and practice don’t always match up, do they?
The reality for a lot of those with hypermobility and EDS has been frustrating, to put it mildly. The 2017 criteria have been criticised widely, too stringent in some areas, too vague in others, and simply not capturing the full picture of how hEDS and HSD actually look in real people. Research has shown that an average of 10.39 years passes between when symptoms begin and when someone receives an accurate hEDS diagnosis, with patients picking up an average of 10.45 incorrect diagnoses along the way [1]. That’s not a minor inconvenience. For many people, that decade of being dismissed, misdiagnosed, and left without proper care has lasting consequences.
So the news that the EDS classification is being updated, properly updated, not just tweaked at the edges, is a big deal. The Ehlers-Danlos Society calls it the Road to 2026, and the new framework is due to be published on 1 December 2026 in the American Journal of Medical Genetics. It will be the first major revision since 2017, and it’s being built on the back of some genuinely important science.
Now, I want to be clear about what this article is and what it isn’t. This is not a prediction piece. I’m not going to tell you exactly what the new criteria will say, because the honest answer is that nobody outside the committee knows that yet. What I can do is explain where we are, what the evidence says, what the researchers have actually found so far, and what the realistic possibilities look like going forward. I’ll also address the question that seems to make a lot of people anxious: will I lose my diagnosis?
This is a long article. It covers a lot of ground, the history of the criteria, the specific research studies feeding into the 2026 update, the preliminary findings already presented, and what it all means practically for those with hypermobility and EDS. If you’re ready to get into the detail, let’s go.
Why the 2017 Criteria Needed Reviewing
To understand why the Road to 2026 matters, you first need to appreciate where the 2017 classification came from, and why it’s proved so complicated in practice.
Before 2017, EDS was classified differently. The Villefranche criteria, introduced in 1997, were the main framework. They were broader, less specific, and by 2017 the scientific consensus was that they were capturing too wide a group of people. The goal of the new classification was to tighten things up, to create a more homogeneous, researchable cohort of hEDS patients. The criteria were made more stringent deliberately, to improve specificity.
And here’s where things got messy.
When the GoodHope EDS clinic in Toronto tested the 2017 criteria against their existing patient database, they found that only 15% of patients who had previously been diagnosed with hEDS under the Villefranche criteria actually met the new 2017 criteria [4]. Most of their highly symptomatic patients were left without a diagnosis of hEDS. They now sat under the newly coined umbrella of “hypermobility spectrum disorder”, a category that, as the Toronto team noted, many healthcare systems don’t formally recognise, which can make accessing appropriate care significantly harder.
The tightening also created problems with the Beighton Score, which is the main tool used to assess generalised joint hypermobility when it comes to the diagnostic process for EDS and HSD. The Toronto study found that Beighton scores assessed by primary care practitioners were higher than scores assessed by EDS specialists in 81% of cases [4]. In other words, the same person could get very different scores depending on who was measuring them. Not exactly the solid, objective foundation you’d want for a diagnostic tool.
A retrospective study from an Italian reference centre, which assessed 327 patients from 213 families who had been diagnosed under the older criteria, found that the 2017 criteria failed to adequately capture the multisystemic nature of hEDS, and proposed that they should be made less stringent to include more patients currently pushed into the HSD category [3]. This isn’t a niche finding, it’s a pattern that has appeared across multiple clinical settings in multiple countries.
And then there’s the gender issue. Over 70% of people diagnosed with hEDS and HSD are female. A 2025 retrospective review of 429 patients found that a staggering 94.4% had been told, by physicians who were not psychiatrists, mind you, that their symptoms were psychological in origin: that they were “making it up”, “attention-seeking”, or suffering from conversion disorder [2]. Only 5.6% had made it to an hEDS diagnosis without passing through some form of psychiatric misdiagnosis. When psychiatric misdiagnosis occurred, the diagnostic delay was substantially longer.
A separate survey of 152 women with EDS in Australia found that more than half first noticed symptoms more than 15 years before they received a diagnosis, and more than three-quarters received at least one other diagnosis first [7]. Let me be blunt here: a diagnostic framework that makes it easier for doctors to dismiss very real physical symptoms as psychological problems is not functioning as it should. That’s one of the things the 2026 update is trying to fix.
What Is the Road to 2026?
The Road to 2026 is an initiative led by a committee from the International Consortium on the Ehlers-Danlos Syndromes and Hypermobility Spectrum Disorders (IC). The committee includes leading experts across clinical practice and research, geneticists, rheumatologists, physiotherapists, and others, as well as community representatives who bring lived experience to the table.
There are three main things the committee is working on:
- An update to the classification criteria, a comprehensive review and revision of the 2017 framework
- A diagnostic pathway, a practical, clinically tested guide to help healthcare professionals (including those who aren’t EDS specialists) diagnose EDS and HSD more accurately
- Assessment and treatment pathways, practical guidance on how to assess and manage symptoms and comorbidities across the spectrum
The final publication will come out in two special issues of the American Journal of Medical Genetics, the first on 1 December 2026 covering the new classification, and a second in early 2027 covering care and management pathways. Both will be open access, which is worth noting, this isn’t going to be locked behind a paywall. Patients and clinicians alike will be able to read it directly.
The committee has been meeting regularly since April 2024, with sessions in New York, Philadelphia, London, Antwerp, and Manchester. At each meeting they’ve been reviewing research, analysing survey data, and working through the evidence base systematically. The committee is also running a Delphi study, a research methodology that uses multiple rounds of questionnaires to reach expert consensus on areas where healthcare professionals agree and where they don’t. Clinicians from around the world have been feeding into this process.
In September 2025, the committee came together with the broader scientific community at the 2025 International Scientific Symposium in Toronto, where preliminary findings from the key research studies were presented and debated. We’ll get to what came out of that in a moment.
The HEDGE Study: Looking for the Gene (or Genes)
Of all the research projects feeding into the Road to 2026, the one that’s generated the most excitement, and the most anxiety in equal measure, is the HEDGE Study. HEDGE stands for Hypermobile Ehlers-Danlos Genetic Evaluation, and it represents the largest ever genetic study of hEDS.
The study enrolled 1,000 people from 86 countries who met the 2017 criteria for hEDS, plus 45 with HSD. All 1,021 viable samples underwent whole-genome sequencing at the Broad Institute in Boston. The goal was straightforward: find the genetic cause (or causes) of hEDS.
Because here’s the thing, and this is fundamental to understanding why getting an EDS diagnosis is so complicated, hEDS is the only one of the 13 Ehlers-Danlos subtypes that has no identified genetic marker. Every other type of EDS can be confirmed via genetic testing. With hEDS, diagnosis is purely clinical, which is why all those problems with the Beighton Score and inconsistent criteria matter so much. Without a gene to point to, you’re entirely reliant on clinical observation, and clinical observation varies enormously between practitioners.
So what did HEDGE find? The honest answer, as of early 2026, is: it’s complicated.
The analysis team has confirmed that fewer than 1% of the 1,045 study participants had genetic variants linked to a different connective tissue condition, such as classical EDS, Marfan syndrome, or Loeys-Dietz syndrome. Those participants have been contacted directly and guided towards appropriate follow-up. For the remaining 99%+, the original clinical diagnosis of hEDS was correct, which is actually important and reassuring news. It confirms that the 2017 criteria, when applied well by experienced clinicians, are capturing the right patients.
However, and this is the bit that’s harder to hear, no single gene has been identified that explains hEDS or HSD. The findings point towards multiple rare genetic variants and complex interactions, rather than a single clear genetic cause. An earlier publication from a different research group had suggested a candidate gene called KLK15 might be linked to hEDS. The HEDGE data found that KLK15 variants were not more common in HEDGE participants than in controls. This doesn’t necessarily rule KLK genes out entirely, but it does suggest KLK15 is not a universal cause of hEDS.
The team is still working through rare variant analysis, looking at uncommon genetic changes that might explain subsets of hEDS, and genotype-phenotype studies, which try to map how genetic variants relate to different symptom patterns. The first publications from HEDGE are expected in 2025 or early 2026, with more to follow.
I know that for a lot of people, the news that “we haven’t found the gene yet” is disappointing. But I’d encourage a more measured read of it. hEDS is increasingly looking like it might be not one condition but a group of related conditions that produce a similar clinical picture. This fits with what many geneticists have suspected for years, that the hEDS phenotype could arise from many distinct genetic variants, and that the condition we’ve been calling hEDS may eventually be split into several more defined subtypes. That’s actually how science tends to work when it comes to complex conditions, it gets more complicated before it gets simpler.
The hEDS/HSD Criteria Review Study
Running alongside HEDGE is a second major study that’s directly relevant to how those with hypermobility are diagnosed in clinical settings. The hEDS/HSD Criteria Review Study is an international research effort led by the hEDS/HSD Working Group of the IC, and it’s doing something the 2017 criteria never went through: systematic, prospective clinical testing.
The study recruited 326 participants across eight international clinical sites, in Canada, the USA, Mexico, the UK, Belgium, and New Zealand. Participants were divided into three groups: those diagnosed with hEDS under the 2017 criteria, those diagnosed with HSD, and controls with other non-hypermobile chronic pain conditions. That last group is crucial, to know what’s specific to hEDS and HSD, you need something to compare against.
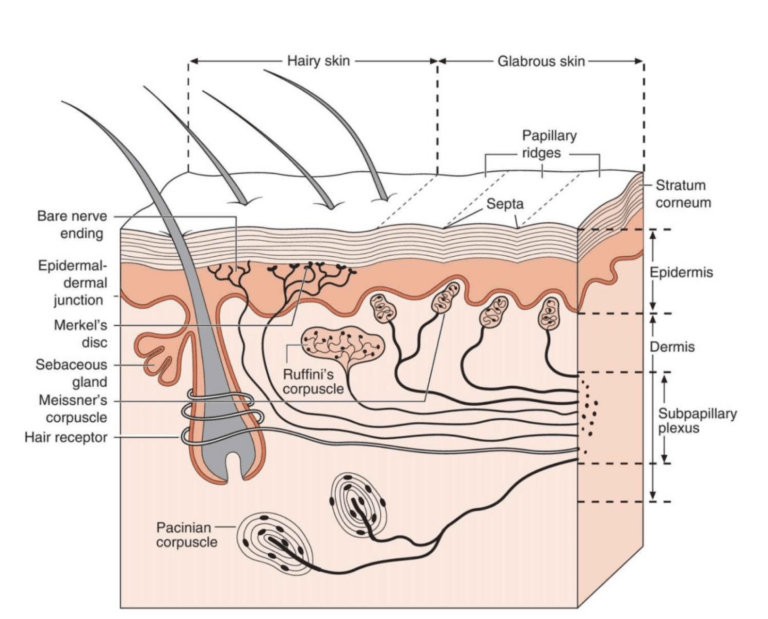
What was measured goes significantly beyond the current Beighton Score. The study collected data on the Beighton Score plus four additional joint assessments: shoulder flexion, forearm rotation, ankle dorsiflexion, and big toe (first MTP) dorsiflexion. It also assessed musculoskeletal, skin, cardiovascular, neurological, gastrointestinal, urogenital, and psychological features, with comorbidities evaluated using validated frameworks including the Rome IV criteria for gastrointestinal conditions.
The statistical approach was rigorous, univariate analysis, latent class analysis, logistic regression modelling, and machine learning were all applied to identify which combinations of variables most strongly predict hEDS/HSD.
Preliminary findings were presented at the 2025 International Scientific Symposium in Toronto. There are a few headlines worth pulling out.
First, the analysis identified new variable combinations that successfully separated individuals with hEDS/HSD from controls. One notable example was the presence of three or more of four specific skin-related parameters, which strongly predicted an hEDS/HSD classification. This is interesting because skin features are currently part of the 2017 criteria but arguably not weighted heavily enough, practitioners often overlook the skin when they’re focused on hypermobility scores. Those of you who’ve read about the full range of EDS symptoms will know that skin involvement, velvety texture, easy bruising, stretch marks appearing early, slow healing, is part of the picture for many people.
Second, and this is a really significant finding, no single measure or variable clearly distinguishes hEDS from HSD. The two conditions appear to exist on a shared biological spectrum with overlapping features. This supports what many clinicians have been saying for years: the hEDS/HSD distinction, as currently drawn, may not reflect a meaningful biological difference. They may be different expressions of the same underlying problem.
Third, the research team plans to test both an expanded 4-joint hypermobility assessment and a proposed revised diagnostic model in a second study that began in late 2025, in real-world clinical practice. So the work is continuing, and the 2026 criteria will be informed by that real-world testing, not just laboratory analysis.
For those with hypermobility who’ve struggled to get an HSD diagnosis taken seriously, this is potentially very meaningful. If the research confirms that hEDS and HSD sit on a spectrum rather than being two distinct categories, the updated framework may reflect that, which could have real implications for how much clinical weight an HSD diagnosis carries. This connects directly to the broader question of what can be confused with hypermobility and why clear diagnostic criteria matter so much.
The Biomarker Picture: Is a Blood Test on the Horizon?
One of the most talked-about recent findings in the EDS research space is the possibility of a blood test for hEDS and HSD. This sounds almost too good to be true, so let’s be precise about what the evidence actually shows.
A study published in the American Journal of Medical Genetics Part A investigated something called extracellular matrix (ECM) fragments in patient plasma [6]. The extracellular matrix is essentially the structural scaffolding of connective tissue, the network of proteins, including collagen, that holds things together. If that scaffolding is being broken down abnormally, traces of the breakdown products should show up in the bloodstream. And if you’re wondering about the collagen question more broadly, we’ve written before about whether those with hypermobility actually have less collagen, it’s worth reading alongside this.
The researchers found that patients with hEDS and HSD showed a distinctive shared pattern of fibronectin and type I collagen fragments in their plasma, a pattern that wasn’t seen in controls or in patients with rheumatoid arthritis, psoriatic arthritis, or osteoarthritis. Importantly, hEDS and HSD showed the same pattern, which the authors argue further supports their classification as a single disorder rather than two separate conditions [6].
This is genuinely exciting preliminary evidence. However, and this is important, we’re not talking about a diagnostic test that exists or is available anywhere. This is laboratory research. The samples were analysed using Western blotting, which is a specialist research technique, not a routine clinical blood test. The findings need to be validated in larger cohorts before anyone can say with confidence that this could form the basis of a clinical diagnostic tool. What it does offer is a potential biological mechanism and a potential direction for future testing.
The genetics research tells a similar story. A 2025 review of diagnostic challenges in hEDS found that among patients who met the 2017 criteria for hEDS, genetic testing revealed an alternative or additional diagnosis in approximately 26% of cases, roughly one in four [5]. This was in a specialist genetics clinic setting, which is important context, the figure may be lower in the general population. But it highlights why researchers argue that genetic testing should become more standard in the hEDS diagnostic pathway, even in the absence of a specific hEDS gene, because it can identify conditions that require different management.
What Might Change in 2026?
This is the question everyone is asking, and I want to be careful here because nobody outside the Road to 2026 committee has seen the final proposals. What I can do is outline what the evidence points towards and what the committee has indicated it’s examining.
The Beighton Score Is Likely to Change
The most consistent theme across the research is that the Beighton Score, in its current form, is not fit for purpose as the primary measure of generalised joint hypermobility. Its scope is narrow, it assesses nine specific joints and gives a maximum score of 9. It doesn’t assess the shoulders, ankles, forearms, or numerous other joints that can be hypermobile. It also doesn’t account for hypermobility that has reduced over time due to age, surgery, or exercise, which is a major problem for older patients with EDS whose scores may no longer reflect their underlying condition.
Now, I’ve written quite a bit about the Beighton Score before in our post on why it matters more than it should, and I’ll be honest, this finding from the Criteria Review Study is exactly the kind of evidence that piece was anticipating. The score has always been a blunt tool. The hEDS/HSD Criteria Review Study has trialled an expanded 4-joint assessment adding shoulder flexion, forearm rotation, ankle dorsiflexion, and first MTP dorsiflexion to the Beighton protocol. Whether this makes it into the final criteria remains to be seen, but the evidence base for expanding beyond the Beighton Score is solid.
The hEDS/HSD Distinction May Be Reconsidered
As mentioned above, the preliminary data suggest that hEDS and HSD share a biological foundation rather than being genuinely distinct conditions. If the 2026 criteria reflect this, moving towards a spectrum model rather than a binary distinction, it could significantly change how HSD is perceived and managed clinically.
For those with hypermobility currently carrying an HSD diagnosis, this would be a positive development. It would mean that the condition you have is recognised as part of the same clinical picture as hEDS, rather than a lower-tier category that many healthcare systems don’t formally acknowledge. This could have implications for access to care, insurance coverage in some countries, and the credibility of the diagnosis in clinical settings. It also speaks to the broader question of how hypermobility presents differently in different people, and why a rigid binary has never been the right model.
Comorbidities and Systemic Features May Get More Weight
One of the strongest criticisms of the 2017 criteria is that they don’t adequately capture the multisystemic nature of hEDS. Conditions like POTS, MCAS, pelvic floor dysfunction, gastrointestinal dysmotility, and others are extremely common in people with hEDS and HSD. Yet they feature very little in the current diagnostic criteria.
The Criteria Review Study assessed these features systematically for the first time. Finding that certain skin-related features strongly predict hEDS/HSD classification is one example of how comorbidities and associated features might be given more diagnostic weight in the updated framework. For those dealing with the full spectrum of EDS-related conditions, whether that’s anxiety, brain fog, or the kind of widespread fatigue that disrupts daily life, having these formally acknowledged in the diagnostic criteria would be a meaningful step.
A Clearer Diagnostic Pathway for Non-Specialists
One of the consistent themes across the research is that most healthcare professionals, GPs, physiotherapists, general rheumatologists, simply aren’t familiar enough with hEDS and HSD to diagnose them reliably. The Road to 2026 is developing a diagnostic pathway specifically designed to help clinicians who aren’t EDS specialists navigate the diagnosis, including potentially an interactive digital tool.
This matters enormously when it comes to diagnostic delay. One of the reasons so many people wait over a decade for a diagnosis is that the clinicians they see first have never encountered hEDS, or have heard of it but aren’t confident applying the criteria. A well-designed clinical pathway could dramatically reduce that delay for future patients. We’ve written before about the impact of medical trauma on those with EDS, and a lot of that trauma stems directly from years of diagnostic wandering.
Possible New Subtypes
As the genetics research matures, there’s a possibility that some patients currently diagnosed with hEDS will receive more specific diagnoses as genetic causes are identified. The HEDGE study’s findings suggest that hEDS may eventually be subdivided into distinct genetic subtypes. This isn’t confirmed, it depends on what further genetic analysis reveals, but it’s a direction that multiple experts in the field have flagged as likely in the long term.
Will People Lose Their Diagnosis?
I want to address this directly because it’s the source of a huge amount of anxiety in the community, and I think people deserve a straight answer rather than vague reassurance.
The short answer is: based on everything we currently know, it’s very unlikely that large numbers of people will have existing diagnoses invalidated.
Here’s why. The HEDGE study showed that fewer than 1% of its 1,000 hEDS participants had a genetic variant suggesting a different condition entirely. The vast majority had correctly diagnosed hEDS under the 2017 criteria. The hEDS/HSD Criteria Review Study’s preliminary findings suggest the relationship between hEDS and HSD is more of a spectrum than a clear boundary, which, if anything, points towards an expansion of diagnostic recognition rather than a narrowing of it.
The Road to 2026 committee has also been clear that its goals include reducing diagnostic delays and improving inclusivity for diverse presentations. It’s not working to make the criteria more restrictive, if anything, the evidence points in the direction of making them more inclusive and more practical to apply.
That said, it’s worth being honest about what a criteria revision could mean in practice. If you received your diagnosis from someone who applied the current criteria loosely, or from a clinician who isn’t a specialist, it’s possible that under any new set of criteria your diagnosis would need to be reconfirmed. This isn’t an attack on your experience or your suffering, it’s just the nature of how medical criteria work. And importantly: the aim of the review is better recognition, not less recognition.
The EDS Society has been clear that the community survey results will feed directly into the published work alongside the scientific findings. Patient experience isn’t an afterthought here, it’s part of the evidence base.
The Community Survey: Your Voice in the Science
In March 2025, the EDS Society launched a comprehensive Community Experience Survey to capture the real-world experiences of people with EDS and HSD. The results of this survey are being shared with the Road to 2026 Scientific Committee and will be published as a paper in the same special issue of the American Journal of Medical Genetics as the classification update itself.
This is genuinely significant. It means that the experiences of patients, access to care, diagnostic experiences, what comorbidities affect day-to-day life, what support is and isn’t available, will sit alongside the scientific evidence in the peer-reviewed literature. Community experience isn’t being gathered and then set aside. It’s being integrated.
The EDS Society had previously run a “Your Voice Matters” survey in April 2024, available in nine languages, to collect community questions, hopes, and concerns about the Road to 2026. That feedback was collated and passed directly to the committee. The process has been, by the standards of medical classification reviews, unusually inclusive of patient voices from the start.
What the Toronto Symposium Showed Us
The 2025 International Scientific Symposium in Toronto, held from 17-21 September 2025, was a hybrid event bringing together researchers, clinicians, and community members. It was the midpoint of the Road to 2026 process, the point at which the committee shared interim findings and engaged with the wider scientific community before moving into the final stages of the classification work.
The preliminary findings from the hEDS/HSD Criteria Review Study, which I’ve outlined above, were presented there. The HEDGE team also gave updates on where the genetic analysis stood. What came across clearly from the symposium coverage is that this is rigorous, ongoing work, not a rubber-stamping exercise. The committee is genuinely wrestling with difficult questions about where the diagnostic boundaries should sit, what the evidence supports, and how to make criteria that work in real clinical practice rather than just in specialist centres.
That process is important. Classification criteria that don’t work in general practice don’t help anyone, regardless of how good the science behind them is.
What Does This Mean If You’re Already Diagnosed?
If you already have an hEDS or HSD diagnosis, the most likely outcome of the 2026 update is that your diagnosis becomes better supported, not undermined. The direction of travel in the research is towards more inclusive criteria, a clearer pathway for clinicians, and a better-validated distinction (or fusion) of hEDS and HSD.
In practical terms, the update is unlikely to change your rehabilitation programme or your day-to-day management significantly, at least not immediately. The fundamentals of exercise and rehabilitation for those with hypermobility and EDS are grounded in physiology, not diagnostic criteria. Building strength and stability, improving proprioception, managing energy, none of that changes based on a classification update.
What the update may change is the quality of care you receive from healthcare professionals who aren’t EDS specialists. A clearer diagnostic pathway and better clinical guidance should mean fewer experiences of being sent away with a shrug. That’s the goal, anyway. In the meantime, approaches to pacing and energy management remain just as relevant as ever for day-to-day function.
What Does This Mean If You’re Still Waiting for a Diagnosis?
This is where I think the Road to 2026 has the potential to make the most meaningful difference in people’s lives.
If you’ve been experiencing the symptoms of EDS and hypermobility, widespread joint pain, frequent flare-ups, skin features, POTS, fatigue, brain fog, but haven’t yet received a diagnosis, the 2026 update may make that process easier in a few ways.
More inclusive criteria could mean that patients currently falling into the “not quite hEDS” gap get a diagnosis that properly reflects their condition. A clearer diagnostic pathway could mean your GP is better equipped to identify whether you need a specialist referral. And a better-validated HSD framework could mean that even if you don’t meet the full hEDS criteria, the HSD diagnosis carries more clinical weight and opens more doors to appropriate care.
None of this will happen overnight. Even after the criteria are published in December 2026, it will take time for them to filter into clinical practice, for guidelines to be updated, and for training materials to be developed. But the direction of travel is genuinely positive. If you’re currently trying to navigate the diagnostic process, our post on what can be confused with hypermobility may be a helpful companion resource for your conversations with healthcare professionals.
What This Doesn’t Change
I want to be honest about something, because I think it’s important not to oversell what a classification update can do.
The diagnostic criteria, however well-designed, are only as useful as the clinicians applying them. The fundamental problem isn’t just that the criteria are imperfect, it’s that most clinicians have never heard of hEDS, aren’t confident diagnosing it, and often dismiss complex, multisystemic presentations as psychological rather than physical. No set of criteria changes that without accompanying education, training, and cultural change in medicine.
The research on psychiatric misdiagnosis is sobering. A 2025 retrospective review found that 94.4% of hEDS patients had been misdiagnosed with psychiatric conditions before receiving their correct diagnosis [2]. That’s not primarily a criteria problem. It’s a medical culture problem. Better criteria will help, but they won’t solve that alone. We’ve written at length about the psychological impact of medical trauma in EDS, and it’s worth reading if this resonates with your own experience.
Likewise, and I say this gently, a diagnosis is the beginning of a management process, not the end of one. The 2026 update will also be developing assessment and treatment pathways, and that second publication, due in early 2027, may be just as important as the classification criteria for people’s day-to-day lives. Understanding what helps, proper rehabilitation, symptom management, addressing comorbidities, is where most of the practical difference is made. That’s where our approach at The Fibro Guy studios sits: working with people on building function and reducing the impact of their condition, wherever they are in the diagnostic process.
A Note on the Genetic Research in Context
I realise I’ve spent a lot of this article on genetics, and I want to make sure I’m not leaving people with the wrong impression.
The search for the genetic cause of hEDS matters enormously for the future, for more accurate diagnosis, for understanding mechanisms, and ultimately for developing targeted treatments. But even if those genes are found tomorrow, it would still take years to translate that into a routine diagnostic test and then into treatments. We’re not at the beginning of that process, but we’re not near the end either.
What I can say with confidence is that the HEDGE study has already produced something valuable: confirmation that the vast majority of people diagnosed with hEDS under the 2017 criteria have been correctly diagnosed. Given the widespread doubt and medical dismissal that many in this community have experienced, and the question of why so many people with hypermobility also have other diagnoses like autism that compound the dismissal, having the world’s largest ever genetic study confirm that the diagnosis is valid is not nothing.
I’ve worked with a lot of people with hypermobility and EDS over the years. One of the most consistent themes is the struggle to be believed, by doctors, by employers, by family members. Having that confirmation carries real weight while the science continues to develop.
The Practical Timeline
So where does all of this land, practically speaking? Here’s the picture as it stands in early 2026:
- The Road to 2026 committee has been meeting since April 2024, reviewing literature, running the Delphi study, and integrating findings from the HEDGE and Criteria Review studies.
- The hEDS/HSD Criteria Review Study is testing a revised diagnostic model in clinical practice settings, with a second study that began at the end of 2025.
- HEDGE publications are expected in 2025-2026, with additional papers to follow.
- The new classification will be published on 1 December 2026 in two special issues of the American Journal of Medical Genetics.
- A second publication covering care and management pathways is planned for early 2027.
- The EDS Society will produce accessible, multilingual resources for patients and clinicians based on the findings, including, potentially, an interactive digital diagnostic tool.
There are still several months to go before the December 2026 publication date. The committee is still working. The science is still being tested. We’ll update this post as significant new information becomes available.
What to Do in the Meantime
If you’re waiting for a diagnosis, I wouldn’t advise waiting for the 2026 criteria before seeking one. The current 2017 criteria are still in use, and a diagnosis under those criteria is valid, it won’t be swept away by the update. Getting assessed now, if possible, gets you access to appropriate care sooner.
If you’re already diagnosed, the most useful thing you can do is exactly what you’re probably already doing: managing your condition as effectively as possible. That means building appropriate strength and stability, understanding your pain mechanisms, addressing comorbidities like POTS and MCAS where present, and working with clinicians who take the condition seriously.
If you haven’t already, it’s worth exploring whether compression garments might help with any dysautonomia symptoms, and looking at what dietary factors might be relevant for your situation. And if sleep is a struggle, which it often is for those with hypermobility, that post is one of our most detailed evidence-based resources.
For practitioners reading this: the case for improving your knowledge of hEDS and HSD before December 2026 is strong. The updated criteria will come with educational resources, but getting ahead of the curve now will serve your patients better. Our comprehensive EDS guide is a reasonable starting point for clinicians who want to understand how we approach the condition from a rehabilitation standpoint.
Frequently Asked Questions
Based on the evidence so far, it is very unlikely that existing hEDS diagnoses will be invalidated. The HEDGE study confirmed that fewer than 1% of participants had a different underlying condition. The direction of the 2026 update appears to be towards more inclusive criteria and a better-validated spectrum approach to hEDS and HSD, not a narrowing of who qualifies for a diagnosis. However, the full criteria have not yet been published, so the precise implications won’t be known until December 2026.
HEDGE stands for Hypermobile Ehlers-Danlos Genetic Evaluation. It is the largest ever genetic study of hEDS, sequencing the DNA of 1,000 people with hEDS and 45 with HSD from 86 countries. As of early 2026, the key findings are: the original clinical diagnoses were correct in over 99% of participants; no single gene has been identified that causes hEDS; the condition appears to involve multiple rare genetic variants and complex interactions; and the KLK15 gene previously suggested as a candidate was not confirmed as a universal cause. First publications from HEDGE are expected in 2025 and 2026.
The new classification framework is scheduled to be published on 1 December 2026 in a special issue of the American Journal of Medical Genetics. A second publication covering care and management pathways is planned for early 2027. Both will be open access, meaning anyone can read them without a subscription.
Several changes are being explored by the Road to 2026 committee. The Beighton Score is likely to be revised or supplemented with additional joint assessments. The hEDS/HSD distinction may be reconsidered in light of evidence that they exist on a shared biological spectrum. Comorbidities and systemic features, including skin findings, POTS, and gastrointestinal features, may receive more diagnostic weight. A clearer diagnostic pathway for non-specialist clinicians is also being developed. None of these changes are confirmed until the December 2026 publication.
Not yet. A study published in the American Journal of Medical Genetics Part A found a distinctive extracellular matrix fragmentation pattern in the plasma of hEDS and HSD patients, pointing towards a potential future biomarker. However, this is laboratory research, the findings need validation in larger cohorts before any clinical blood test could be developed. A routine diagnostic blood test for hEDS does not currently exist.
References
[1] Halverson, C.M., Cao, S., Perkins, S.M., Francomano, C.A. (2023) ‘Comorbidity, misdiagnoses, and the diagnostic odyssey in patients with hypermobile Ehlers-Danlos syndrome’, Genetics in Medicine Open, 1(1), 100812. doi: 10.1016/j.gimo.2023.100812 [2] Lee, C., Chopra, P. (2025) ‘The Incidence of Misdiagnosis in Patients with Ehlers-Danlos Syndrome’, Children, 12(6), 698. doi: 10.3390/children12060698 [3] Ritelli, M., Chiarelli, N., Cinquina, V., Vezzoli, M., Venturini, M., Colombi, M. (2024) ‘Looking back and beyond the 2017 diagnostic criteria for hypermobile Ehlers-Danlos syndrome: A retrospective cross-sectional study from an Italian reference center’, American Journal of Medical Genetics Part A, 194(2), pp. 174-194. doi: 10.1002/ajmg.a.63426 [4] McGillis, L., Mittal, N., Santa Mina, D., So, J. et al. (2020) ‘Utilization of the 2017 diagnostic criteria for hEDS by the Toronto GoodHope Ehlers-Danlos syndrome clinic: A retrospective review’, American Journal of Medical Genetics Part A, 182(3), pp. 484-492. doi: 10.1002/ajmg.a.61459 [5] Forghani, I., See, J., McGonigle, W.C. (2025) ‘Hypermobile Ehlers-Danlos Syndrome: Diagnostic Challenges and the Role of Genetic Testing’, Genes, 16(5), 530. doi: 10.3390/genes16050530 [6] Ritelli, M., Chiarelli, N., Cinquina, V., Bertini, V. et al. (2025) ‘Bridging the Diagnostic Gap for Hypermobile Ehlers-Danlos Syndrome and Hypermobility Spectrum Disorders: Evidence of a Common Extracellular Matrix Fragmentation Pattern in Patient Plasma as a Potential Biomarker’, American Journal of Medical Genetics Part A, 197(1). doi: 10.1002/ajmg.a.63857 [7] Trudgian, R., Flood, T. (2024) ‘An exploration of the journey to diagnosis of Ehlers-Danlos Syndrome (EDS) for women living in Australia’, PLOS ONE, 19(7), e0307574. doi: 10.1371/journal.pone.0307574, The Fibro Guy Team ,